The First Week Progress Report
Introduction: There have been more than a week since I came to Boston. Many things happened in the first week. With the help of Professor Gu and Tutor Kim , I have completely understand the schedule and the aims in different stages. Furthermore I solve some problems under the guidance.
Keywords: Adaption , Preparation
Progress Description: I had read the review “Coming of age : ten years of next-generation sequencing technologies” before I arrived in Boston. Then I read the review “Genetic sources of population epigenomic variation”.Those two reviews gave me abstract of recent researches on NGS technique and population epigenomic variation, and I am trying to find combinations of them. I met some problems in understanding the papers especially the methods on researching.
Examples of problems in “Genetic sources of population epigenomic variation”, Nature reviews, 2016 ;Volume 17.
- Page 324-325 “In humans, the majority(70–80%) of all CpG dinucleotides are methylated across tissues, with unmethylated CpGs being mainly confined to CpG islands in promoter-proximal regions.” According to what I know, when CpG islands are methylated , it may lead to several promoters being methylated at the same time. Does it have connections with the appearance of tumour. Or, in another word, I want to know more about methylated CpG islands which result in tumor suppressor gene’s inactivation of transcription.
- Page 326 “ This lack of variation is not only visible within populations but also between human populations.” I think there is something described not clearly enough. What does the former “population” actually represent? Should “human populations” be included in “ populations”?
- Page 330 “ These challenges stem mainly from the fact that differential methylation states (epialleles) can be inherited meiotically for many generations, so that detected cis associations are simply a reflection of LD between sequence alleles and epialleles rather than the result of any type of regulatory interactions.” The author didn’t explain “LD” beside it. So I was wondering what “LD” represents at first.Tutor Kim pointed that LD is linkage disequilibrium and recommended me not to ignore figures or boxes in papers.She also taught me to turn to GEO if I might want to learn more, which is a good suggestion for me.
Next Stage Work: I haven’t decided the title of my presentation,but I already came across an idea: Inspired by population genetics of 5mC in humans, I am interested in the difference between array based researches and NGS based researched researched. Since I also read the review on NGS, maybe I can find the connections and make it the subject. Moreover , on page 326 “ They interrogated ~3.2 million CpG sites throughout the genome, 15% of which could be associated with meQTL. This is about a two to threefold increase compared to results from array based studies, suggesting that genetic effects are much more prevalent than previously appreciated.”Maybe I can find out the reason , make a hypothesis and design experiments to confirm it.


(波士顿儿童医院研究所) (导师的医院兼研究所)
来BOS的小建议如下:
- 商店,抵达波士顿后第一个去的商店推荐star market,波士顿地区比较大的购物中心,一是商品齐全(包括水果、蔬菜、肉类、熟食、零食、饮品),二是可以在资讯台购买交通卡(7天卡、15天卡或30天卡,轻轨、地铁、公交无限次通用)。
- 出行,主要有步行、公交(包括地铁、轻轨、公交车等)、出租车。步行:如果不需要通过查尔斯河,步行是可行的,如需过河就会很麻烦。公交:性价比很高,波士顿的公交网分布很广,四通八达,(但是公交卡只能在地下地铁站或star market购买,地下地铁站只有在市中心才有)。出租车:推荐用uber等软件,价格很贵,起步价10美元左右,再加小费,除非赶时间,不推荐。
- 热水,美国的水龙头里的热水不建议直接喝,并且一般商店里很少卖电热水壶,可以自己带一个小的电热水壶来。
- 气候,夏季日照时间很长(早5时至晚9时),温度最高的时间是下午3时至5时,温度很高,盛夏约34°C,阳光刺眼,建议出行带太阳伞或太阳帽。
- 洗衣,公寓有洗衣机和烘干机,洗完直接烘干,没有晒衣服的习惯。
The second week progress report
Abstract:
I have read many other papers in this week ,and I changed my title under the guidance of professor and tutor. For the reason that I have known something about Multiple Myeloma (MM) before and I want to do further research to find the connection between MM and DNA methylation. CpG islands methylation analysis can be used for prognosis, and methylation content of genome can be used to judge the malignant degree.
Aim:
- The analysis of candidate gene meythlation in different stages of MM and its connection with the damage of kidney. (MM always results in the loss-function of kidney.)
- The connection between methylation content of genome and MM staging.(It may be a new supporter in MM diagnosis.)
MM stages are under DS staging system,which is divided into Ⅰ、Ⅱ、Ⅲ .The function of kidney is judged by erum creatinine and divided into A(normal) and B(above normal) .(A: serum creatinine<2.0 mg/dL, B :serum creatinine>2.0mg/dL)
Candidate genes:
SOCS-1, p16,E-caherin, DAP kiniase, p73, MGMT, p15, RARβ
Samples:
- MM patients (massive,different in gender/paraprotein isotype/stage),their bone marrow (BM) and peripheral blood (PB).
- MM cell lines
- Patients with nonmetastatic solid tumors,their BM samples for comparison.
- Healthy volunteer,normal PB samples.
Steps for analysis of 8 candidate gene meythlation:MSP
- Isolate genomic DNA from cell lines and patient samples in standard methods.
- Use methylation-specfic PCR to do qualitative analysis.
- Separate on 2.5% agarose gel elecrophoresis.
- Ethidium bromide staining.
MSP:(1)NaHSO3 treatment of genomic DNA.(change unmethylated C into U) (2)Use MSPprimers to design two different primer pairs that are specific for either the methylated or unmethylated sequence.(3)PCR process:Reactions are hot-started at 95°C for 5 min and held at 80°C before addition of 0.625 U of Taq polymerase.35 cycles of 95°C for 30 s, 60°C for 30 s and 72°C for 30 s, followed by one cycle of 72°C for 5 min. (Primer pairs:C in unmethylated primers turns into T will lead to lower G+C ,and results in lower Tm temperature)
Results:
- MSP result;(example)
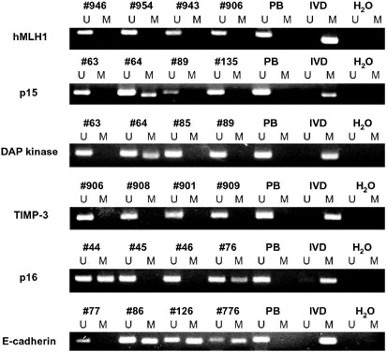
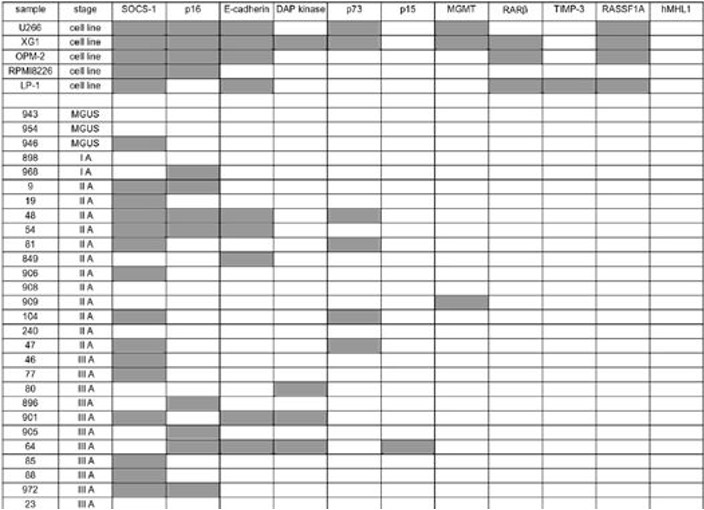
- Make the qualitative analysis of 8 candidate gene methylation in different stages of MM .(Ⅰ、Ⅱ、Ⅲ)
- Make the qualitative analysis of 8 candidate gene methylation depends on the function of kidney. Do assessment on if each of candidate gene has remarkable influences by two-sided fisher exact or chi-square test, it depends.(example)
Steps for analysis of methylation content of genome:HPLC
- Isolate genomic DNA from samples.
- Use HCl or HF to make DNA into bases.
- Let the product pass through chromatographic column.
- Compare the results and determine the absorption peak and its quantity by ultraviolet light.
- Calculate the integral area of 5mC and get the methylation level of the whole genome.
Results:
Use tables and charts to make connections between the methylation level of the whole genome and MM staging.


